Protein-ligand docking is an essential method in computer-aided drug design and structural bioinformatics. It can be used to identify active compounds and reveal molecular mechanisms of biological processes. A successful docking usually requires thorough conformation sampling and scoring, which are computationally expensive and difficult. Recent studies demonstrated that it can be beneficial to docking with the guidance of existing similar co-crystal structures. In this work, we developed a protein-ligand docking method, named FitDock, which fits initial conformation to the given template using a hierachical multi-feature alignment approach, subsequently explores the possible conformations, and finally outputs refined docking poses. In our comprehensive benchmark tests, FitDock showed 40%~60% improvement in terms of docking success rate and an order of magnitude faster over popular docking methods, if template structures exist (> 0.5 ligand similarity). FitDock has been implemented in a user-friendly program, which could serve as a convenient tool for drug design and molecular mechanism exploration.
FitDock is a command-line application running on Linux or Windows systems.
We also developed a graphical user interface plugin for docking with PyMOL.
The program, PyMOL plugin and datasets are freely available for academic users here . Other users please contact us.
| Alignment | Refinement |
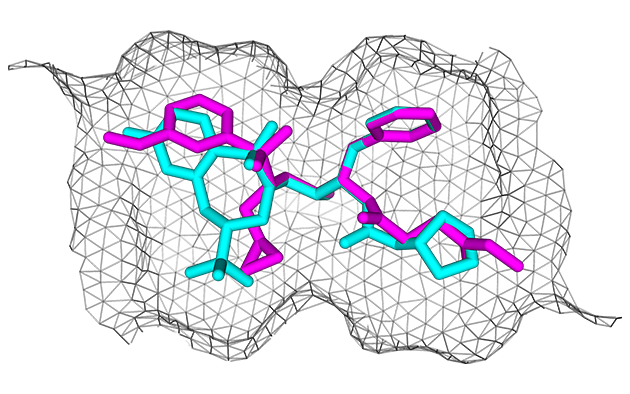 |
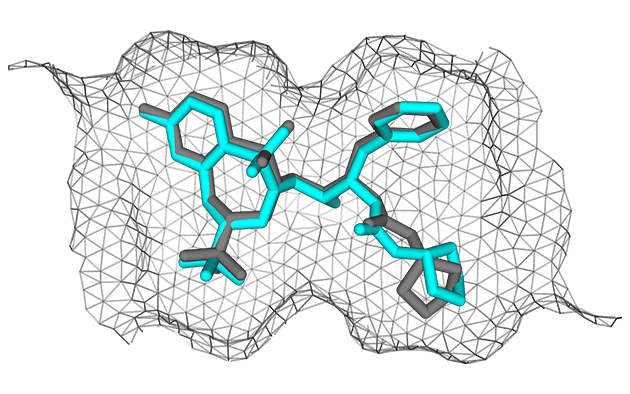 |
Publication
Xiaocong Yang, Yang Liu, Jianhong Gan, Zhi-Xiong Xiao, Yang Cao. FitDock: improved protein-ligand docking with template fitting. Briefings in Bioinformatics. 23(3), 2022, https://doi.org/10.1093/bib/bbac087
Youjun Wang, Yuchan Yang, Yang Liu, Zhi-Xiong Xiao, Yang Cao. FitDockApp: a Graphical User Interface Plugin for Template-based Docking With PyMOL,Progress in Biochemistry and Biophysics. 51(3):716-725, 2024