
Help
Quick Links
• How it works• Submitting a job
• Check the job results
• Chemical Library
• Contacts
• Issues
• References
How it works?
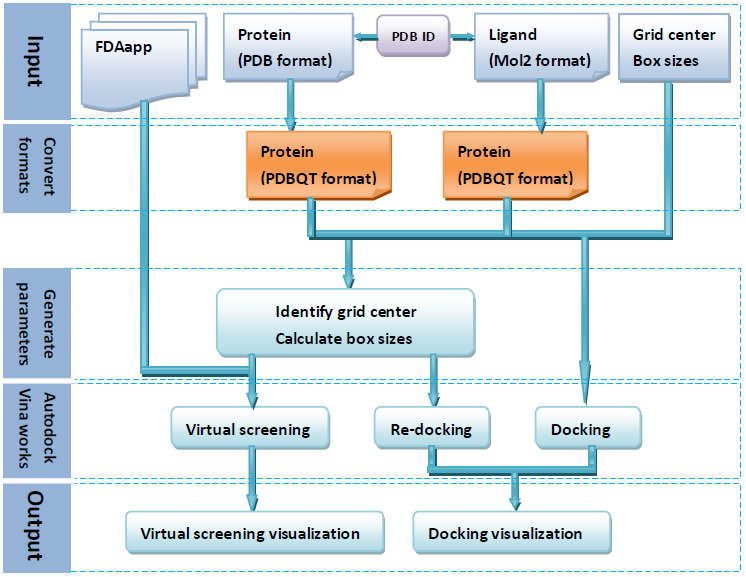
The web sever can identify automatically the binding site using AutoDockTools and calculate the docking box sizes using eBoxSize after users upload the structure file of protein receptor and the structure file of its ligand or a molecule binding to the binding site of the receptor. There are many (11908) protein structures and the structures of their lignads which are obtained from PDBbind database deposited in the web server. Users only need to enter a PDB ID to complete a virtual screening. The docking progarm of the web server is Autodock Vina which has been commonly used in virtual screening. The following figure is workflow diagram of the web server.
Submitting a job
DrugScreen provides users two services: Virtual Screening and Docking.
Virtual Screening
For virtual screening, the user must enter Job ID, User, Output Number. If a protein receptor has a small molecule ligand in PDB, users only need to enter a PDB ID of the protein receptor. If a protein receptor has no ligand in PDB, users can also enter the Protein and a Known Ligand to this target. There are some input rules:
• Job Id : A special ID for a virtual screening.
• User : User name. The user must remember his/her user name which will be used to check the virtual screening results.
• Output Number : The number of output molecules
• Protein (PDB ID) : The PDB ID of the protein receptor. If you want to know whether a protein receptor has a ligand, you can
search the information of the protein receptor in PDBbind.
• Protein : The protein receptor. The protein receptor is uploaded in PDB format, and all HETATMs and all solvent molecules should be
removed and all hydrogen atoms should be added before uploading.
• Known Ligand to this target : The ligand of receptor or a molecule which can bind to the binding site of receptor and be used to
find the binding site of receptor automatically. The ligand is uploaded in MOL2 format and all hydrogen atoms should be added
before uploading.
• The symbol "-" can't appear in the name of an uploaded file or the input. For example, a file should not be uploaded as "ab-cd.pdb".
If you still don't know how to submit a job, the Screen page has a Example and you can see the Iuput of the example.
Docking
The docking service includes normal Docking and Re-docking. There are some input rules:
• For normal Docking, center_x, center_y and center_z are 3 Autodock Vina parameters and stand for 3 coordinates of the binding site center.
size_x, size_y and size_z are also 3 Autodock Vina parameters and stand for docking box sizes. These parameters can be set using
AutoDockTools.
• For normal Docking, Protein is uploaded in PDB format, and all HETATMs and all solvent molecules should be removed and all hydrogen
atoms should be added before uploading. Ligand is uploaded in MOL2 format and all hydrogen atoms should be added before uploading.
• Re-docking is the docking of the native ligand and the receptor in the crystal structure of a complex.
• The input rules of Re-docking are same as Docking.
• The symbol "-" can't appear in the name of an uploaded file or the input. For example, a file should not be uploaded as "ab-cd.pdb".
If you still don't know how to submit a job, the Dock page has a Example and you can see the Iuput of the example.
Check the job results
When a virtual screening or docking job is completed, the result will be visualized using 3Dmol or JSmol.
• The docked molecule can be viewed and downloaded in docking result page without logging in.
• For a virtual screening result, the user can view the top 30 docked molecules and download the output molecules after registration and
logging in.
• If the user registers successfully, he/she can log in and check the result. The user may receive a registration confirmation e-mail. In order to
receiving e-mails successfully, try to use academic or non-profit mailboxes.
Chemical Library
DrugScreen provides users with a chemical library FDAapp containing 1806 FDA-approved drugs. These compounds of the library are from DrugBank database and each of these compounds has a DrugBank ID. Users can use these DrugBank IDs of these compounds to serch for them from DrugBank.
Contacts
If you have any problems with DrugScreen, please contact us at:
• Jixiang Liu : liujixiang2016@yeah.net
• Yang Cao : cy_scu@yeah.net
Issues
1. The server can run in most browsers, but there are some problems when it runs in IE. If you can't open DrugScreen in your browser, you had
better use Firefox or Google Chrome to open it.
2. The symbol "-" can't appears in the name of a uploaded file or the input. For example, a file should not be uploaded as "ab-cd.pdb".
References
1. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility.
Journal of Computational Chemistry, 2009, 30(16): 2785-2791
2. Feinstein WP, Brylinski M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted
binding pockets. Journal of Cheminformatics, 2015, 7: 18
3. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and
multithreading. Journal of Computational Chemistry, 2010, 31(2): 455-461
4. Rego N, Koes D. 3Dmol.js: molecular visualization with WebGL. Bioinformatics. 2015, 31(8): 1322-1324
5. Hanson RM, Prilusky J, Renjian Z, et al. JSmol and the next-generation web-based representation of 3D molecular structure as applied to
Proteopedia. Israel Journal of Chemistry, 2013, 53(3-4): 207-216
6. Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Research,
2006, 34(Database issue): D668-D672
7. Renxiao W, Xueliang F, Yipin L, et al. The PDBbind Database: Collection of Binding Affinities for Protein-Ligand Complexes with Known
Three-Dimensional Structures. Journal of Medicinal Chemistry, 2004, 47(12): 2977-2980