
How it works
DrugRep provides users two services: Receptor-based Screenand Ligand-based Screen.
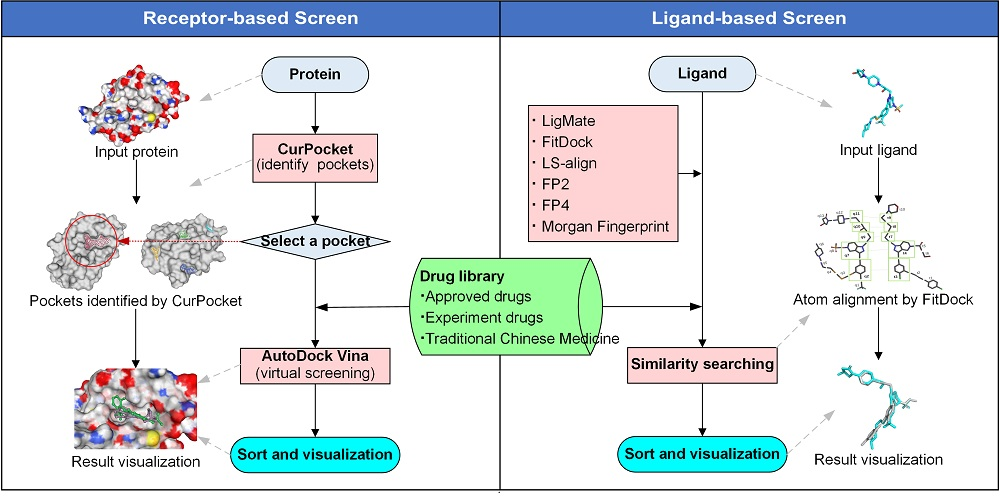
Receptor-based Screen
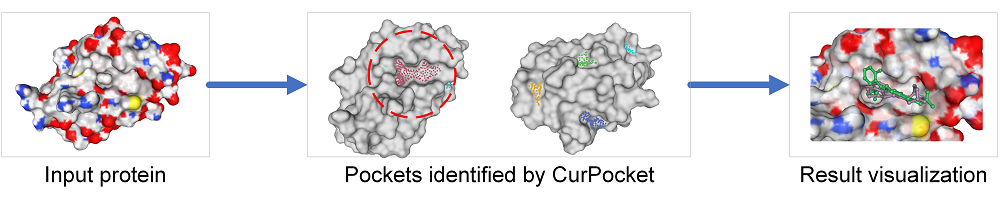
Receptor-based Screen can identify the binding pockets and docking parameters including the grid center coordinates and the sizes of the search space using CurPocket, a curvature-based cavity detection approach issued by Cao Lab. In addition, users also can upload ligand that co-crystallize with the receptor for calculating the docking pocket's grid center and box sizes based on AutoDockTools and eBoxSize. Receptor-based Screen then perform batching docking over drug library using AutoDock Vina. In Receptor-based Screen, users only need to enter a PDB ID or to upload the structure file of a protein receptor to inspect pockets, and then select a pocket to screen after inspecting.
Ligand-based Screen
Ligand-based Screen offers ligand-similarity based virtual screening methods. For a given active ligand, it can search for compounds with different types of similarity, including LigMate , FitDock-align Morgan Fingerprint , LS-align , FP2 and FP4 .
1. LigMate : LigMate developed by our lab is a multi-feature integration algorithm, which can be applied to ligand-similarity-based virtual screening. These descriptors of LigMate focus on the maximum common substructures (MCSS), the relative distance of atoms from the ligand mass center (ILDS), as well as the ring differences (RS).
2. FitDock-align : FitDock is a novel template-based docking method, which fits initial conformation to the given template using a hierachical multi-feature alignment approach, subsequently explores the possible conformations. DrugRep only adopts the alignment approach to find equivalent atom pairs between the two compounds. PC-score here was used to measure the superposition of the two molecules.
3. Morgan Fingerprint : Morgan Fingerprints, a circular fingerprint, is also a topological fingerprint. Morgan Fingerprints are obtained by modifying the standard Morgan algorithm. This can be roughly equivalent to Extended Connectivity Fingerprints (ECFPS). The web server calculates the similarity between ligands using the RDKIT package.
4. LS-align : LS-align is an algorithm designed for atom-level structural comparison of ligand molecules. The target function of LS-align is a combination of inter-atom distance, atom mass, and chemical bond connections; while the final atom-to-atom alignment is generated by maximizing such target function through an enhanced-greedy based, iterative heuristic search algorithm. LS-align program contains two modules: Rigid-LS-align for rigid-body ligand structure comparison; and Flexi-LS-align for flixible structure comparison. In particular, the Flexi-LS-align module seeks for optimal alignments of various alternative conformers of the ligand molecules by rotating flexible bond-angles, which allows the consideration of binding-induced conformational changes in the ligand structural comparison and alignment.
5. FP2 : A path-based fingerprint which indexes small molecule fragments based on linear segments of up to 7 atoms (somewhat similar to the Daylight fingerprints):
- A molecule structure is analysed to identify linear fragments of length from 1-7 atoms. Single atom fragments of C, N, and O are ignored. A fragment is terminated when the atoms form a ring.
- For each of these fragments the atoms, bonding and whether they constitute a complete ring is recorded and saved in a set so that there is only one of each fragment type. Chemically identical versions, (i.e. ones with the atoms listed in reverse order and rings listed starting at different atoms) are identified and only a single canonical fragment is retained.
- Each remaining fragment is assigned a hash number from 0 to 1020 which is used to set a bit in a 1024 bit vector
6. FP4 : FP4 uses a series of SMARTS queries stored in SMARTS_InteLigand.txt
Submitting a job
DrugRep provides users two services: Receptor-based Screen and Ligand-based Screen.
Receptor-based Screen
Receptor-based Screen requires the inputs of PDB ID or Protein Structure File, Pockets Number before inspecting pockets. It also requires the inputs of Pocket, Library, Output Number and Email before screening.
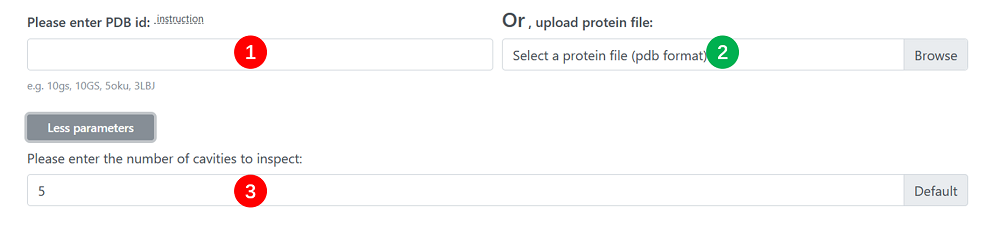
1. PDB ID : Enter a four-length PDB id containing letters and numbers (e.g. 10gs, 10GS, 5oku, 3LBJ ). If it exists in the PDBbind database, you do not need to enter the second parameter, protein structure file. If it does not exist, you need to enter the second parameter. The parameter is case non-sensitive and alternative.
2. Protein Structure File : The protein receptor is uploaded in PDB format, and all HETATMs and all solvent molecules should be removed and all hydrogen atoms should be added before uploading. If the first parameter is not entered, the parameter is required.
3. Pockets Number : The number of pockets. The range of this number is 0 to 20, the default is 5 which can be adjusted.
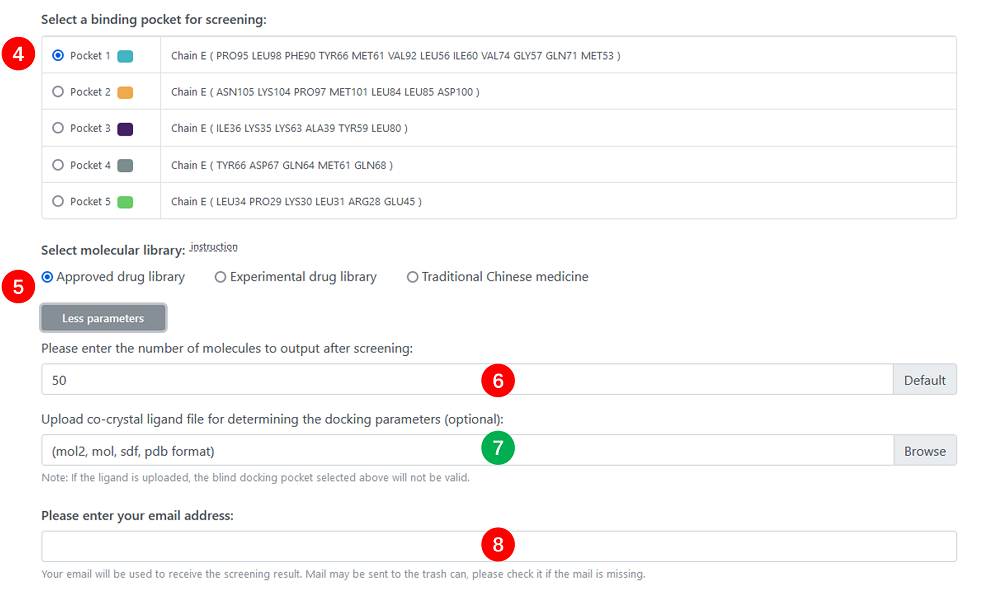
4. Pocket : Select a pocket from inspecting-pockets'result.
5. Library : Ligand molecular library.
6. Output Number : The number of output molecules. The range of this number is 0 to 50, the default is 50 which can be adjusted.
7. Ligand File : This is an optional option for uploading the ligand file that co-crystallizes with the receptor protein. If the user uploads the ligand file, the pocket parameters will be calculated based on the 3D position and size of the ligand, and the pocket selected with the fourth parameter will be invalid. If the uploaded file is incorrect (for example, the ligand and receptor do not originate from a co-crystalline complex), the pocket chosen by the fourth parameter will be used. The ligand is uploaded in MOL2, MOL, SDF, PDB format with 3d structure and all hydrogen atoms should be added before uploading.
8. Email : The screening work may cost several hours. So you can check the result in your mailbox after a period of time instead of waiting on this page. You don't need to enter this parameter if you are a registered user.
Ligand-based Screen
For Ligand-based Screen, the user needs to enter Ligand Structure File, Library, Output Number.

1. Ligand Structure File : The ligand is uploaded in MOL2, MOL, SDF format.
2. Library : Ligand molecular library.
3. Method : Similarity searching methods.
4. Output Number : The number of output molecules. The range of this number is 0 to 50, the default is 10 which can be adjusted.
5. Email : Your email will be used to receive the screening result. You don't need to enter this parameter if you are a registered user.
Check the job results
The especially Receptor-based Screen work may cost several hours. So you can check the result in your mailbox after a period of time instead of waiting on this page. If you are a registered user, you can click your user name in navigation bar to get access to your records.
When a virtual screening is completed, the result can be viewed with abundant information, such as 3D interactive view (using NGL), 2D images, the number of hydrogen bond donors, the number of hydrogen bond acceptors, and the number of rotatable bonds, logP and so on.
Chemical Library
DrugRep offers three drug libraries, including Approved drug library, Experimental drug library and Traditional Chinese Medicine. The first two are from DrugBank database (version 5.1.7); Traditional Chinese Medicine library is from Topscience Company. The latter contains 2390 monomer compounds from nearly 800 traditional Chinese medicines, including various structural types such as flavonoids, alkaloids, terpenoids, and glycosides. Traditional Chinese Medicine library is a powerful library in the fields of anti-cancer, anti-inflammatory, antibacterial, apoptosis, and autophagy research.
RDKit and Open Babel are used to construct 3D conformation for drugs without 3D structure. Open Babel is used to convert file formats, including pdbqt format for receptor-based virtual screening, sdf, mol, mol2 format for ligand-based virtual screening, svg format for viewing.
DrugRep deposits the structure files of 11908 protein receptors and their ligands obtained from PDBbind database. So that users can choose to enter only the PDB ID to complete a virtual screening job for Receptor-based Screen.
Download drug IDs report:
Contacts
If you have any problems with DrugRep, please contact us at:
- Yang Cao : cy_scu@yeah.net
FAQs
-
Browser Compatibility
We tested DrugRep using the latest versions of IE, Edge, Firefox, Chrome and Safari. Browser compatibility is shown in the table below. If you are using one of the before mentioned browsers and you have problems displaying the “Results” page, we recommend you to update your browser to the latest version. Some browsers may not support WebGL or do not support all WebGL features needed by NGL viewer like Opera. Please try another browser in the case you get a warning that WebGL is not supported or the example provided by us on the “Results” page is not visible.
OS Chrome(v96) Firefox(v88)
Firefox(v88) Edge(v96)
Edge(v96) Safari
Safari
Linux (ubuntu18) Windows10 MacOS -
Why can't I receive the result email ?
The mail may be identified as spam. You may need to check in the trash can and add our account to the whitelist. We will to send you an email only after you submit your job. In addition, you can save your jobs by registering and logging in, so you only need to click on your username in the upper right corner to view your jobs.
-
Why does the url in the results email not show the results ?
Your submission may have expired as the server only saves the results for limited days.
Download
We provide a command-line interface for RBS (the receptor-based virtual screening) for Linux.
If you need our software, please register or contact cy_scu@yeah.net
References
- Gan J, Liu J, Liu Y, Chen S, Dai WT, Xiao ZX, Cao Y. DrugRep: an automatic virtual screening server for drug repurposing. Acta Pharmacol Sin. 2022.
- Liu Y, Grimm M, Dai WT, Hou MC, Xiao ZX, Cao Y. CB-Dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol Sin. 2020;41:138-44.
- Liu Y, Yang X, Gan J, Chen S, Xiao ZX, Cao Y. CB-Dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022;50:W159-64.
- Yang X, Liu Y, Gan J, Xiao ZX, Cao Y. FitDock: protein-ligand docking by template fitting. Brief Bioinform. 2022;23:bbac087.
- Grimm M, Liu Y, Yang X, Bu C, Xiao Z, Cao Y. LigMate: a multifeature integration algorithm for ligand-similarity-based virtual screening. J Chem Inf Model. 2020;60:6044-53.
- Cao Y, Li L. Improved protein-ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics. 2014;30(12):1674-80.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785-91.
- Feinstein WP, Brylinski M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J Cheminform. 2015;7:18.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-61.
- Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlic A, Rose PW. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34:3755-8.
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al.. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074-82.
- Wang R, Fang X, Lu Y, Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem. 2004;47:2977-80.
- Hu J, Liu Z, Yu DJ, Zhang Y. LS-align: an atom-level, flexible ligand structural alignment algorithm for high-throughput virtual screening. Bioinformatics. 2018;34:2209-18.